


INTRODUCTION
Interstitial lung diseases (ILDs) comprise an heterogenous group of more than 200 lung disorders characterized by variable amount of fibrotic and/or inflammatory lesions1. The pathogenesis, diagnosis, classification and treatment of ILDs still present a major challenge for clinicians and researchers, due to their appreciable heterogeneity among individuals and their overlapping clinical presentation and radiological patterns. Erroneous radiologic interpretation and delay in management have been associated with significant morbidity and mortality1,2.
An aberrant repair process leading to inflammation and fibrosis with enhanced deposition of collagen in the pulmonary parenchyma is the hallmark of ILD pathogenesis3. The cumulative damage to the pulmonary epithelium results in epithelial cell apoptosis, fibroblast activation and transforming growth factor-beta (TGF-β)-mediated differentiation to myofibroblasts and extracellular matrix remodeling. The aforementioned lead to lung parenchymal destruction and potentially permanent functional impairment4. Although the precise mechanisms have not been fully elucidated, the exposome and the genome of each individual seem to have a cardinal role in a ‘multiple-hits’ pathogenic process4.
Given the progressive nature of these disease entities, early diagnosis is of great importance in order to ensure timely therapeutic intervention and slowdown of disease progression5. Diagnostic workout hinges on thorough clinical evaluation, high-resolution computed tomography (HRCT), pulmonary function testing, laboratory blood testing including serology testing, and importantly multidisciplinary discussion1,2. Current ILD classifications are based on the etiological factors, morphological characteristics and natural history of each disease entity, and patients may be simultaneously classified in more than one category5. Interestingly, diagnosis can be modified over time and sometimes a working diagnosis approach is set in case all potential diagnoses are managed with the same treatment regimen5. Τhe aim of this article is to present developments in the current pharmacological therapies of ILDs and to highlight novel molecular targets and ongoing research trials that may significantly change the prognosis of this group of diseases in the future.
DEVELOPMENTS
Despite extensive research efforts to date, management of ILDs remains challenging. During the last years, the emergence of treatment guidelines6 represented a major step towards attenuating confusion in the field; yet, well-designed trials for novel compounds are still needed6,7. The most promising treatment options and experimental pharmacotherapies for ILDs are presented in Table 1.
Table 1
Treatment options and experimental pharmacotherapies for ILDs
[i] ILD: interstitial lung disease. IPF: idiopathic pulmonary fibrosis. SSc: systematic sclerosis. CYC: cyclophosphamide. MMF: mycophenolate mofetil. Gal: galectin. LPAR1: lysophosphatidic acid receptor 1 antagonist. TGF-β1: transforming growth factor-β1. RA: rheumatoid arthritis. EULAR: European league against rheumatism. ACR: American college of rheumatology. IIMs: idiopathic inflammatory myopathies. ICS: intravenous corticosteroids. MTX: methotrexate. ICS: intravenous corticosteroids. AZA: azathioprine. HP: hypersensitivity pneumonitis. PPF: progressive pulmonary fibrosis.
To this end, the two main treatment regimens in the context of ILDs are anti-inflammatory and antifibrotic compounds. Pharmacological management is driven by lumping ILDs according to the presumptive predominant pathogenic mechanism, although the coexistence of inflammation and fibrosis may be challenging8.
In patients with ILDs and radiologic predominance of inflammatory abnormalities, such as those with connective tissue diseases (CTD-ILDs), hypersensitivity pneumonitis (HP) or non-specific interstitial pneumonia (NSIP), corticosteroids and steroid sparing agents are the mainstay of treatment8-10. However, some patients with stable disease and minor clinical findings, can be closely monitored without receiving any treatment8.
Antifibrotics were initially launched in IPF11. However, the term ‘progressive pulmonary fibrosis’ (PPF) has been recently coined to describe fibrotic lung diseases, other than IPF, that develop into a progressive disease phenotype within a period of 1 year, as defined by clinical, functional and/or radiological parameters6,12. PPF is a descriptive term applied to any fibrotic ILD diagnosis including patients with CTD-ILD, idiopathic NSIP, sarcoidosis, hypersensitivity pneumonitis, occupational ILD and those with otherwise unclassifiable disease6. Based on the fact that pulmonary fibrosis is an end stage situation requiring therapy to hinder disease progression, there has been much interest in whether antifibrotics could be beneficial in PPF6,12.
Idiopathic pulmonary fibrosis
IPF represents a chronic, debilitating and ultimately lethal ILD of unknown origin occurring primarily in older adults, with a highly variable and heterogeneous disease course6,13; yet, the survival has been dramatically improved (almost doubled) with the application of current anti-fibrotic agents. IPF typically affects individuals aged ≥60 years, with most common symptoms being unexplained insidiously worsening dyspnea and dry cough, velcro-type crackles bilaterally and finger clubbing, in the absence of signs and symptoms suggestive of a systemic disease2. Usual interstitial pneumonia (UIP) is the histopathologic and radiologic hallmark of the disease2.
Despite extensive research efforts, no curative drug therapies are available. Pirfenidone and nintedanib are beneficial in patients with IPF, as they decelerate the rate of decline in forced vital capacity (FVC)6,13. Nintedanib, originally designed as an anti-cancer drug, is a tyrosine kinase inhibitor that binds to several growth factor receptors, resulting in attenuation of neo-angiogenesis and inhibition of several pro-fibrotic effects14. In INPULSIS 1 and 2 phase 3 clinical studies, treatment with nintedanib significantly reduced the rate of FVC decline and, therefore, intercepted disease progression over 52 weeks, in comparison with the placebo arm15. Moreover, nintedanib is associated with delayed occurrence of the first acute exacerbation of the disease15. The most frequently reported adverse reaction of nintedanib is gastrointestinal intolerance and specifically diarrhea, which in most cases is manageable15. Pirfenidone is a synthetic pyridine that inhibits fibrotic, inflammatory and oxidation pathways, through downregulation of inflammatory cytokines and pro-fibrotic factors, including TGF-β and tumor necrosis factor-a (TNF-a)11. Four phase 3 trials demonstrated that pirfenidone decelerates the decline in functional indices of patients with IPF. Moreover, meta-analyses showed a decrease in mortality rates in patients with IPF treated with pirfenidone16,17. Adverse effects of pirfenidone include skin reactions, such as rash and photosensitivity and gastrointestinal disorders, which can be reversed if drug dose adjustment or short-term treatment interruption is applied16,17. To date, the combination of pirfenidone and nintedanib, or their co-administration with other non-antifibrotic agents, has not demonstrated further beneficial effects18,19.
With regard to corticosteroids in IPF, their use is strongly discouraged, either as single-agents, or in combination with other anti-inflammatory agents13. Other repurposed drugs proven ineffective or harmful in IPF are endothelin receptor antagonists, interferon gamma, imatinib, everolimus, warfarin and antacids6,13.
Connective tissue disease-ILDs
Except from solely pulmonary entities, ILDs may present as manifestations of autoimmune and connective tissue diseases20. Patients who present with interstitial pneumonia and characteristics of autoimmunity, without a definite diagnosis of a CTD, are classified as interstitial pneumonia with autoimmune features21.
ILD is a main cause of mortality in patients with systemic sclerosis (SSc) and the extent of fibrosis on the HRCT is a major predictive factor of disease mortality7. The latest recommendations for SSc-ILD suggest that cyclophosphamide is implemented in case of disease progression, while mycophenolate mofetil (MMF) use is neither recommended nor discouraged7. However, treatment algorithms according to experts suggest either MMF or cyclophosphamide as agents of choice for induction treatment and MMF for maintenance treatment22. In a randomized trial, MMF presented better toxicity profile and fewer premature treatment withdrawals, as cyclophosphamide has been associated with potential risk of myelosuppression, gonadal and urinary tract toxicity23. In cases of SSc with life-threatening organ involvement, autologous hematopoietic stem cell transplantation is recommended, only in an individualized benefit and risk assessment context7.
A considerable proportion of patients with RA presents with perceptible ILD manifestations throughout their life and the overexpression of mucin 5B has been associated with higher risk24. Patients with severe or moderate to severe active RA and inadequate response to TNF-α antagonist therapy are treated with rituximab in combination with methotrexate25,26. No specific treatment for RA-ILD has been suggested yet. Recently published data from the INBUILD27 and SENSCIS – a cohort of patients with SSc-ILDs – trials showed that nintedanib might be efficacious for patients with CTD-ILDs including RA-ILDs and SSc-ILDs, as it reduced FVC decline; yet, nintedanib was not beneficial for other features of the SSc clinical spectrum28. Finally, the TRAIL 1 study aimed to compare pirfenidone and placebo in patients with RA-ILD. This study seemed to be severely hampered by COVID-19. The primary outcome was not met; yet, patients on the pirfenidone arm presented with a slower decline in FVC. Treatment-related side effects were comparable to other trials29. However, findings must be interpreted cautiously.
ILD is the most severe extra-muscular involvement of the diverse group of idiopathic inflammatory myopathies (IIMs) that is often characterized by the NSIP pattern in histopathology and imaging30. Occasionally, IIM-related ILDs are associated with rapidly progressive respiratory failure presenting as acute respiratory distress syndrome and diffuse alveolar damage30. The presence of myositis-specific autoantibodies may be closely linked to distinct phenotypic characteristics in IIM-related ILDs and may also be decisive for prognosis and treatment30. Anti-aminoacyl-tRNA-synthetase antibody appears to be a favorable prognostic factor, associated with a good response to corticosteroid treatment31, while anti-melanoma differentiation-associated gene-5 antibody is frequently related to high mortality rates, due to rapidly progressive and immunosuppression-refractory disease32. Corticosteroids as monotherapy or combined with anti-inflammatory compounds are considered to be the cornerstone of treatment for ILD associated with IIMs33. Patients with acute or subacute form, defined as respiratory deterioration within 3 months, are typically treated with intravenous pulses of methylprednisolone and subsequent high doses of oral prednisolone34 while cyclophosphamide and rituximab are second-line therapeutic options for refractory cases35,36. Given the disease recurrence, long-term maintenance therapy is indispensable and MMF or azathioprine are the pharmacological agents of choice37.
Hypersensitivity pneumonitis
HP represents a potentially fatal ILD, caused by multiple environmental inhaled antigens triggering exaggerated immunological reactions in susceptible hosts and resulting in inflammation, granulomas and, in some cases, fibrotic lesions38,39. Recent guidelines have subclassified HP as fibrotic and non-fibrotic, a notion with important prognostic and treatment implications39. The key HRCT features of non-fibrotic HP are parenchymal abnormalities and small airway disease, including ground-glass opacities, poorly defined centrilobular nodules and mosaic attenuation or air-trapping40. Typical features of fibrotic HP include traction bronchiectasis, coarse reticulation and volume loss, with or without honeycombing. The coexistence of the aforementioned with mosaic attenuation and diffuse axial distribution differentiate HP from other fibrosing ILDs40.
Given the lack of an established treatment algorithm, therapeutic management of HP is usually based on patient’s clinical features and disease phenotype. The cornerstone of HP treatment is complete avoidance of the causative agent, if identified, in order to prevent disease progression38. If so, most cases of acute HP have a self-limiting course, but a close patient monitoring needs to be ensured38,41. In cases of non-fibrotic HP with features suggestive of persistent active inflammation, corticosteroids and other immunosuppressants should be considered, when important deterioration of pulmonary functional markers or disease progression are present38,41. Fibrotic HP is more likely to respond to antifibrotics and nintedanib has recently been shown to be beneficial in HP when presenting as PPF6,38.
Progressive pulmonary fibrosis
A substantial proportion of patients with several ILDs including HP, RA-ILD and SSc-ILD presents with progressive phenotype. This entity was denominated ‘progressive fibrotic ILDs’, however the new guidelines introduced the term PPF6. The definition of PPF integrates fibrotic ILDs, excluding IPF, that worsen in at least two of the following: respiratory symptoms, functional indices or radiological findings, in twelve months with no alternative explanation6,12. Physiological evidence of disease deterioration includes an absolute change of at least -5 with regard to FVC% predicted or at least -10 to DLCO% predicted within 1 year of followup6,12. These specific thresholds mainly aim to homogenize patients enrolled in clinical trials. New fibrotic changes, such as irregular reticulation, ground glass opacities, traction bronchiectasis, honeycombing and volume loss must be present in HRCT in order to meet the radiological criteria6,12.
Based on the hypothesis that the use of antifibrotics may decelerate PPF worsening, similar to their beneficial effects on IPF, several clinical trials were introduced in order to assess the possible effectiveness of pirfenidone and nintedanib in progressive fibrosis. The INBUILD trial showed that patients with progressive fibrosis under nintedanib treatment had a nearly 50% slower decline in FVC over one year compared to placebo27. The SENSCIS study reported that nintedanib decreased the rate of FVC decline and the subsequent disease progression in patients with SSc-ILD, although it seemed to have a limited role in other manifestations of SSc28. Furthermore, the concomitant use of MMF and nintedanib presented with a good safety profile, with no difference in nintedanib’s treatment effect in this subgroup of patients of the SENSCIS study42. The RELIEF trial suggested that adding pirfenidone to pre-existing treatment in patients with progressive fibrosis may attenuate disease progression, compared to the placebo group43. Of note, RELIEF trial was prematurely terminated due to recruitment issues and thus results should be interpreted with caution. According to the results of the INBUILD trial and the new guidelines, treatment with nintedanib is suggested in PFF, when conventional therapies have failed6,27. Therapeutic decisions should be individualized, based on treatment-related risk and benefit.
Other management concepts and overall care
Comprehensive ILD management extends beyond pharmacological therapy, as non-pharmacological interventions are also essential. Preventative strategies, such as smoking cessation, influenza, pneumococcal, and COVID-19 vaccination and exposure abatement are important and should be recommended to all patients44,45. Patients with severe hypoxemia with symptoms at rest or on exertion should be given oxygen supplementation, as this intervention has a major role in their health-related quality of life46. Pulmonary rehabilitation should also be encouraged, as it is highly effective in patients with chronic lung diseases47.
Proactive assessment and treatment of patient’s comorbidities, such as gastro-esophageal reflux, chronic obstructive pulmonary disease, lung cancer and pulmonary hypertension, can affect disease mortality48,49. Pulmonary hypertension is a severe, underdiagnosed comorbidity of ILDs that adversely impacts quality of life and functional status. Recently, the INCREASE study demonstrated that the inhaled prostacyclin analogue treprostinil significantly improves 6-minute walk distance, an indicator of exercise capacity, decreases N-terminal pro b-type natriuretic peptide levels, a biomarker of cardiac dysfunction, and is associated with fewer disease exacerbations in patients with ILD complicated by pulmonary hypertension compared to placebo50,51.
Given that ILDs are often associated with dramatic restraints in everyday life and survival, palliative care consultation should also be considered timely, with patient-centered interventions52. Lung transplantation remains an intervention that increases life expectancy, thus early referral of rapidly progressors or non-responders to available pharmacotherapies is advised53. Nonetheless, only a small number of candidates meet the eligibility standards, due to disease-specific challenges, such as advanced age and comorbidities53.
Future perspectives
Despite the development of pharmacological agents and the establishment of non-pharmacological interventions with promising therapeutic potential, effective treatment of pulmonary fibrosis is still an unmet need. This mainly stems from the fact that a considerable proportion of patients will still develop progression or will present with acute exacerbations. Additionally, one out of four patients will discontinue antifibrotic therapy.
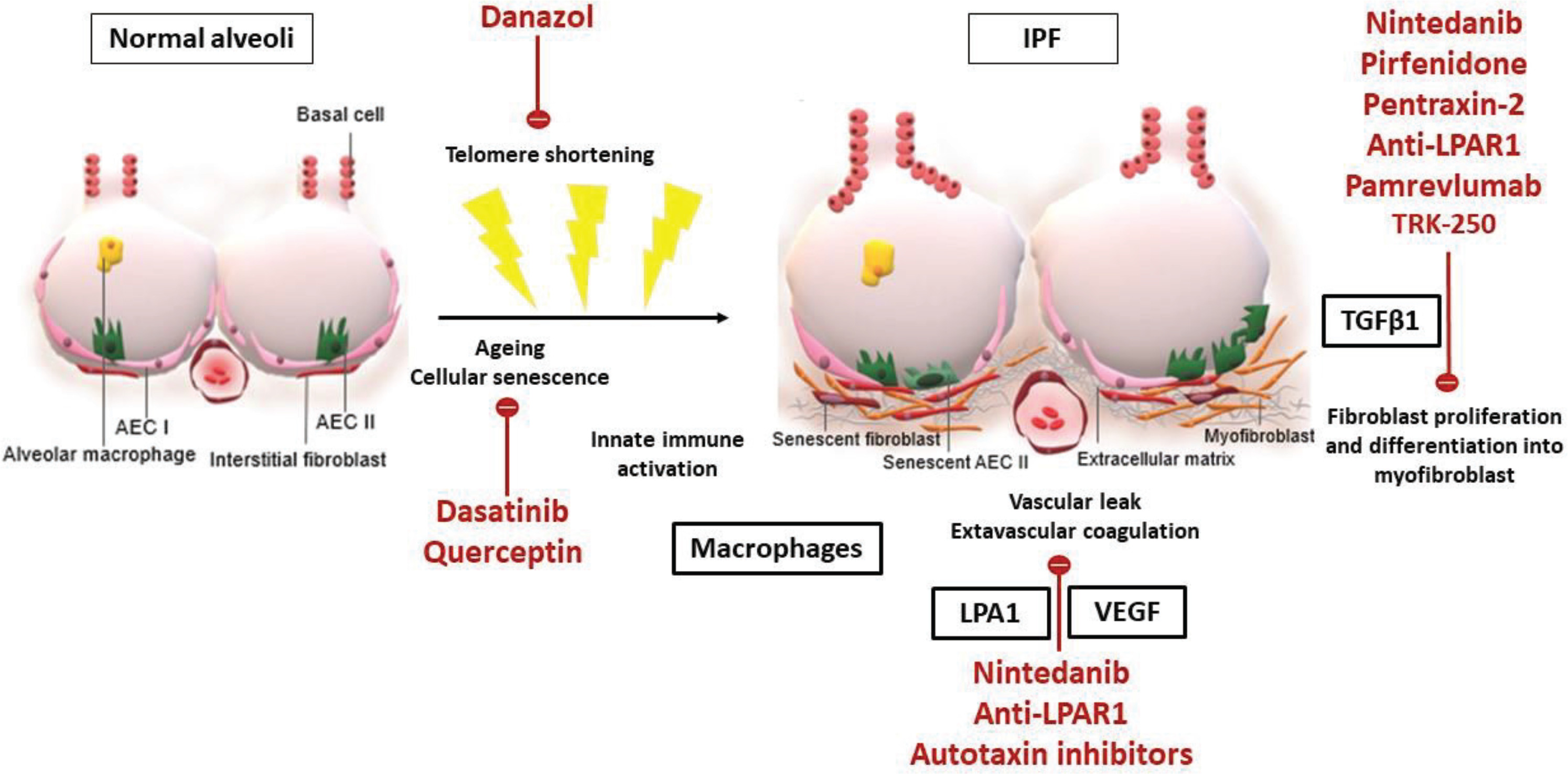
Thus, there is a pressing need for an oncologic approach, with multiple compounds targeting different pathways being administered concomitantly. Current research activity could hopefully broaden the therapeutic horizons of IPF by identifying new pathogenic pathways and potential molecular targets. The main pathophysiologic changes in IPF alveoli and the molecular pathways that current treatment and experimental pharmacotherapies target are shown in Figure 1. Multiple novel agents targeting immune responses or profibrotic processes, such as autologous lung stem cells (NCT04262167, NCT02745184), tRNA synthetase inhibitor (NCT03711162, NCT04888715), TGF-1 suppressor (NCT03727802), lysophosphatidic acid receptor 1 antagonist (NCT04308681), inhaled galectin-3 inhibitor (NCT03832946), nitrogen oxide inhibitors (NCT03865927), serpin peptidase inhibitor (NCT03538301), senolytic combination of dasatinib and quercetin (NCT02874989) are being investigated in clinical trials with promising preliminary results44. The synthetic androgen danazol has demonstrated potential effect on short telomere syndromes and a phase 2 clinical trial, aiming to study its effects in pulmonary fibrosis associated with telomeropathies, is currently in process (NCT04638517)54. Intravenous infusion of recombinant human pentraxin-2 – a protein that inhibits monocytes’ differentiation into macrophages and profibrotic fibrocytes – initially showed promising results55; yet, the phase 3 clinical trial was prematurely terminated due to futility (NCT04552899, NCT04594707). Pamrevlumab – a recombinant human monoclonal antibody targeting connective tissue growth factor and acting as a mediator of signal transduction in fibrogenic processes – is also in phase 3 development for IPF (NCT03955146, NCT04419558), given its promising results in tolerability and efficacy in phase 2 trials56. GLPG1690 (ziritaxestat), which inhibits autotaxin, an enzyme important for generating lysophosphatidic acid, has also demonstrated an encouraging tolerance and effectiveness profile57, that supports further clinical assessment (NCT03733444, NCT03711162). Additionally, a phase 2 clinical study demonstrated safety and efficacy of a PDE4B inhibitor, BI 1015550, in slowing down functional progression of IPF58. Safety profile was acceptable, with diarrhea being the most commonly reported adverse reaction. The most important attribute of the trial was the synergistic effect of PDE4B inhibitor with current standard of care, highlighting the need for an oncologic approach. Efficacy was observed, despite enrolling patients with more progressive disease, indicated by an almost double functional decline in the placebo arm compared to antifibrotic treatment arm (-59.2 mL in 12 weeks) than that reported in the INPULSIS study (-113.6 mL during 52 weeks)59. Considering the role of such compounds in preventing COPD exacerbations and progression of lung fibrosis, a potential benefit for individuals with combined pulmonary fibrosis and emphysema should be investigated in the context of future clinical trials. These are consistently excluded from previous randomized clinical trials; yet, they represent almost 25% of IPF cases seen in the everyday clinical practice and deserve attention.
CONCLUSION
The main challenge of trials of novel compounds is the fact that they should be effective as an add-on therapy, given that novel agents cannot be compared to true placebo. This demands a deep insight into pharmacokinetics, pharmacodynamics and drug-drug interactions. The disappointing results of human pentraxin 2 trial is a typical paradigm showing how demanding is the challenge of launching an add-on therapy. Another challenge is whether some compounds including inhaled pirfenidone and thyroid hormone have potential as inhaled therapy, sparing thus systemic adverse events and concomitantly being effective60,61. Finally, an important challenge is the identification of compounds able to improve quality of life, except drugs able to slow down FVC decline. The extended-release form of the dual-acting ƙ opioid receptor agonist/μ opioid receptor antagonist, nalbuphine, has great potential with regard to chronic cough reduction in patients with pulmonary fibrosis62. Taken together, integration of individually tailored therapeutic regimens as dictated by personalized medicine approaches is of paramount importance. Reproducible theragnostic biomarkers will hopefully revolutionize the treatment landscape for patients living with ILDs.