


INTRODUCTION
Pulmonary alveolar proteinosis (PAP) is a rare syndrome characterized by the accumulation of amorphous lipoproteinaceous material in the alveolar space and alveolar macrophage dysfunction1. Its incidence is 0.2 cases per million inhabitants2. Clinically, it is characterized by hypoxia and increased risk of secondary infections and pulmonary fibrosis, with a variable clinical course ranging from spontaneous resolution to death due to progressive respiratory failure or infections3.
PAP can be classified according to its etiology into primary (90–95% of cases), secondary, and congenital1. Primary PAP (pPAP) is characterized by an interruption in granulocyte-macrophage colony-stimulating factor (GM-CSF) signaling, dividing into autoimmune or hereditary3. In secondary PAP (sPAP) cases, there is a reduction in the functions and/or number of alveolar macrophages, usually secondary to hematological disorders. However, they can also result from malignancy, immunodeficiencies, infections, chronic inflammatory syndromes, or toxic inhalation4. Finally, congenital PAP (cPAP) cases result from gene mutations that participate in the production of surfactant proteins1.
We present the case of a patient diagnosed with PAP associated with adenovirus infection, an unexpected association.
CASE PRESENTATION
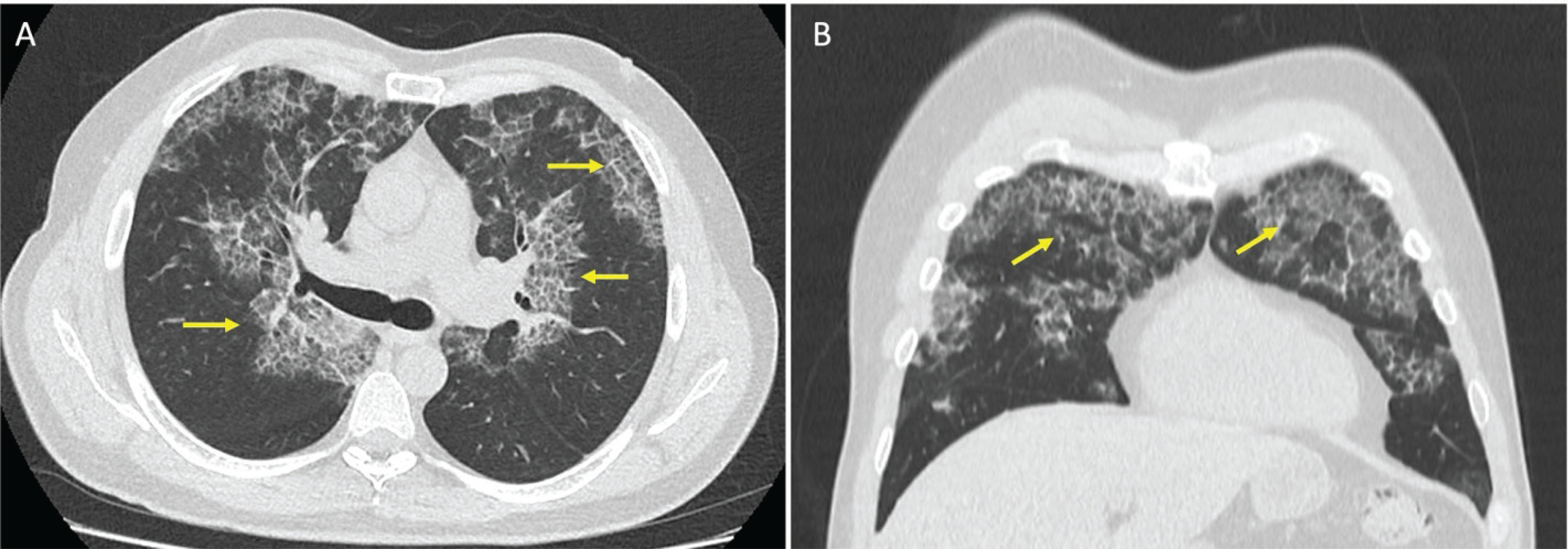
A 53-year-old man visited the emergency department for a 7-day evolution of cough, general malaise, shortness of breath, chills, and low back pain. He was a patient with only a history of smoking for 20 years but no other past medical history. Vital signs on admission showed a temperature of 38.1°C, blood pressure of 135/70 mmHg, heart rate of 95 beats/min, respiratory rate of 22, and oxygen saturation of 84% on room air. Physical examination showed extensive bilateral crackles. Initial laboratory results revealed elevated D-dimer (610 ng/mL, reference value <250 ng/mL) and LDH (323 U/L, reference value 140–280 U/L). Complete blood count, electrolytes, renal function, and liver chemistries were within normal range. An initial chest X-ray showed multilobar pulmonary opacities. Due to the elevated D-dimer, a chest contrast-enhanced computed tomography (CT) was performed. There was no pulmonary embolism but it showed patchy ground-glass opacities with peri broncho vascular distribution, predominantly in the upper lobes, associated with thickening in inter- and intra-lobular septa, with a crazy-paving pattern and ground-glass opacities associated with alveolar occupation in the upper segment of the left lower lobe, forming consolidation foci (Figure 1).
Figure 1
Chest computed tomography (CT) scan. Axial (A) and coronal (B) views with patchy ground-glass opacities with peri broncho vascular distribution associated with thickening in the inter- and intra-lobular septa, consistent with a crazy-paving pattern (yellow arrows)
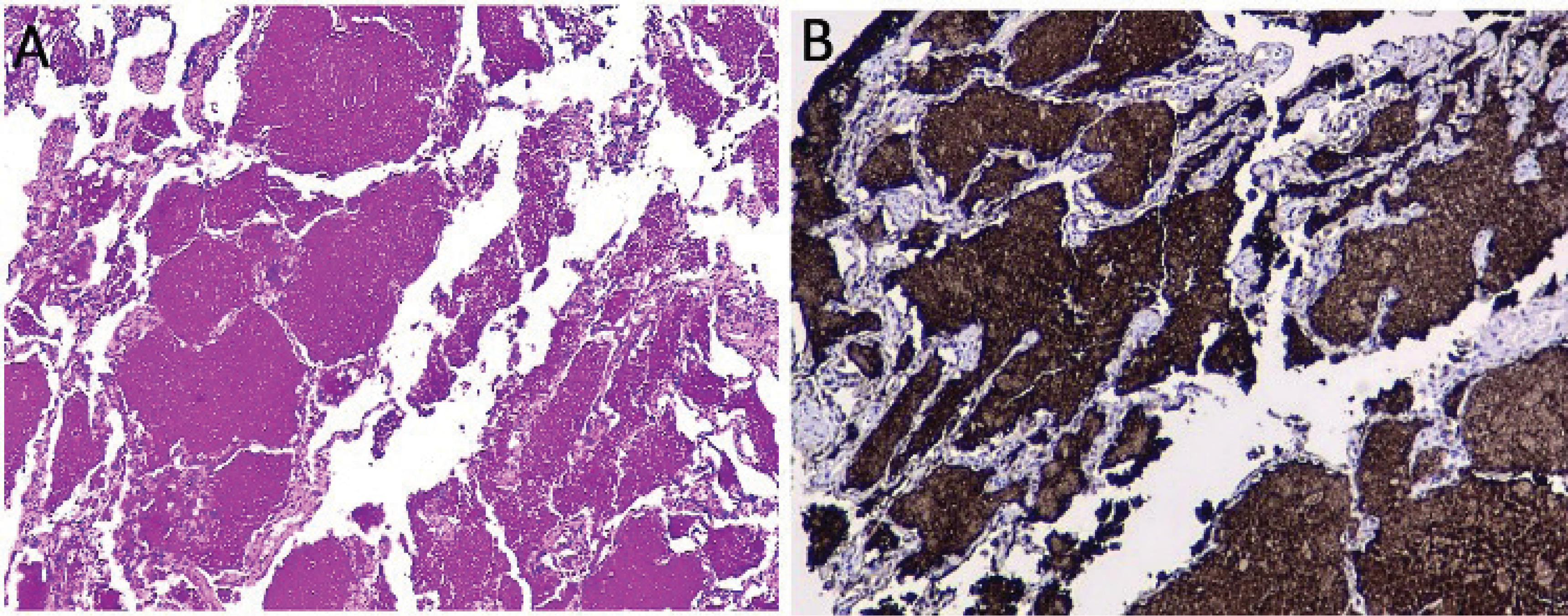
Given the clinical presentation and the radiological pattern, a possible differential diagnosis was organizing pneumonia associated with viral infection with possible bacterial superinfection. A respiratory virus film array was ordered and was negative. It was decided to start steroid (IV methylprednisolone 40 mg daily) and antibiotic management for community-acquired pneumonia pathogens. The patient had temporary improvement for two days; however, the cough worsened, associated with greenish expectoration, and worsening of crackles and wheezing. A bronchoscopy was performed, which revealed abundant yellowish mucus with inflammation of the mucosa in the right and left bronchial tree. A transbronchial biopsy showed a bronchial wall covered by ciliated columnar epithelium on a wall with a slight mononuclear inflammatory infiltrate associated with the interstitial thickening of lung parenchyma. It was evident that the alveoli were occupied by granular proteinaceous material, and in other areas, macrophages loaded with brown pigment and cholesterol crystals were visualized. The proteinaceous material was positive for periodic acid-Schiff (PAS), PAS with diastase, and reagent for surfactant A in the immunohistochemistry study (Figure 2). An alveolar fluid’s film array was positive for adenovirus, with no evidence of fungi, mycobacteria, or other microorganisms. PAP diagnosis was made. Autoimmunity studies (antinuclear antibodies and rheumatoid factor) were performed, along with protein electrophoresis and peripheral blood smear, without revealing any abnormalities.
Figure 2
Transbronchial biopsy. Interstitial thickening of lung parenchyma. Alveoli were occupied by granular proteinaceous material, and in other areas, macrophages loaded with brown pigment and cholesterol crystals. Proteinaceous material stained positive in PAS stain (A) and positive for surfactant protein A (B)
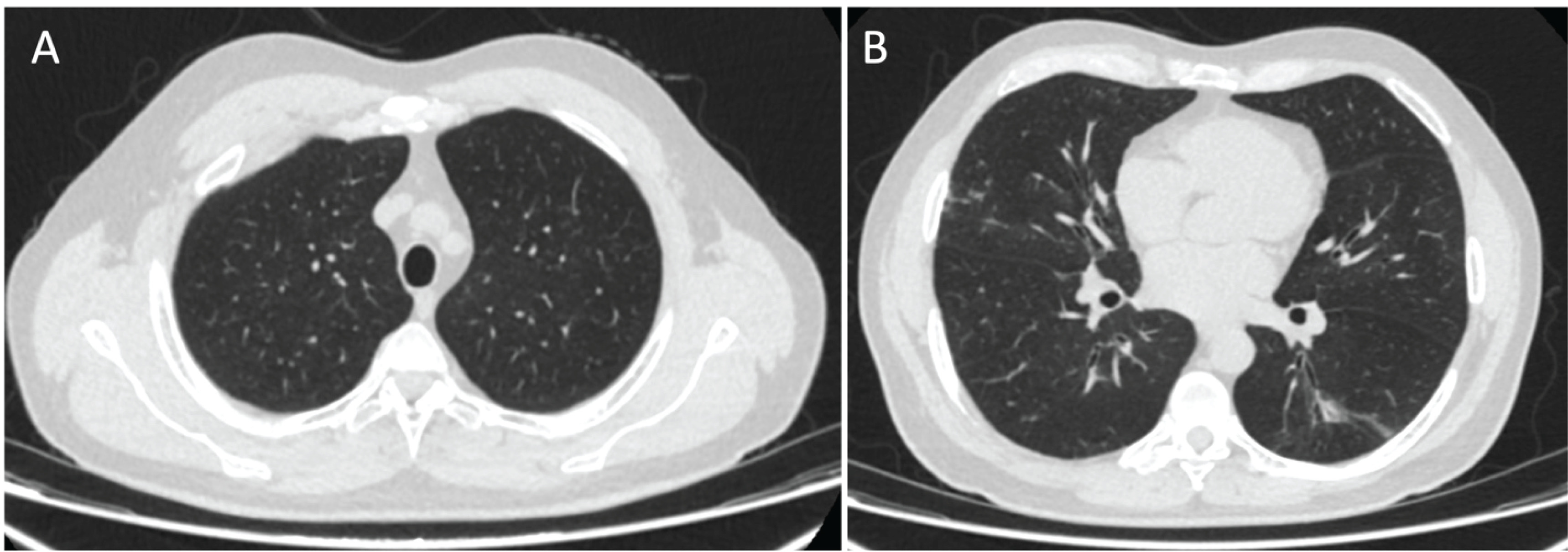
Considering the above, it was proposed that the etiology of alveolar proteinosis was associated with adenovirus infection. Finally, antibiotic management was extended for 14 days with a short course of steroids. The symptoms progressively improved, and the symptoms were relieved after 15 days. The patient was evaluated four months later with no symptoms and resolution of chest CT findings (Figure 3).
DISCUSSION
PAP is a rare disease that usually presents with progressive dyspnea and cough along with fever, pain, and hemoptysis. It is more common in men, with a 2:1 ratio to women and a median age of diagnosis of 50 years5. sPAP cases are <10% of all PAP cases6. PAP can develop after a predisposing factor reduces the number of or affects the function of alveolar macrophages, leading to an impaired ability to remove surfactant from the alveolar epithelium1.
In most cases, the study of sPAP is done through case reports or series. Zhang et al.6 conducted a retrospective analysis of PAP cases, showing that 9 cases corresponded to sPAP. Most patients were men (66.6%), and the median age was 37 years. When analyzing the etiologies, 5 cases had hematological disorders (4 myelodysplastic syndrome and one chronic myelogenous leukemia), and 4 cases had tuberculosis (TB). Similarly, Ishii et al.4 concluded that hematologic disorders are the primary underlying disease in >75% of cases with adult-onset sPAP.
Infections are an important etiological component of PAP cases. There is controversy about whether infection is the disease’s specific cause or consequence. Some authors argue that some microorganisms, especially TB, impair macrophages’ phagocytic function, generating an accumulation of serous surfactant material6. However, it is also known that surfactants proteins contribute to the host’s pulmonary defence through the opsonization of microbial pathogens and direct microbial destruction7. Hence, in cases of PAP, there is a predisposition to become infected with common microorganisms (Streptococcus, Klebsiella, Haemophilus, Staphylococcus, Pseudomonas, Serratia, Proteus, Escherichia) and opportunistic microorganisms (Nocardia, Mycobacterium tuberculosis, Mycobacterium avium-intracellulare, Pneumocystis jirovecii)8. The association between PAP and viral infections, especially influenza, Epstein-Barr, cytomegalovirus, and SARS-CoV-2, has also been reported9,10. To our knowledge, this is the first case report of the association between PAP and adenovirus infection.
Some authors have considered that the presence of anti-GM-CSF antibodies is the cornerstone of diagnosing pPAP (autoimmune); however, there is the possibility that infections lead to the development of antibodies and the development of PAP9. In our case, although we do not have the measurement of anti-GM-CSF antibodies, which is a limitation of our study, we propose that the etiology of PAP was secondary to the adenovirus infection due to the course of the patient’s disease, where he had complete resolution of symptoms after drainage with bronchoscopy and without recurrence of symptoms, which is similar to the self-limited course of a viral infection, in addition to the negativity of autoimmunity markers. Recently, Li et al.11 employed an in vitro cellular model to investigate the functions of non-canonical inflammasomes in macrophages in response to adenovirus infection, and for the first time it was found that adenovirus infection induces the activation of caspase-4 and caspase-5, which subsequently leads to pyroptosis of macrophages, resulting in a decrease in their number in the alveolar epithelium.
The management of PAP depends on the symptoms. In asymptomatic cases or with mild symptoms, it is recommended to perform clinical and radiological follow-up along with a pulmonary function test. However, in moderate to severe symptomatic cases, whole lung lavage or a trial of experimental treatment (subcutaneous or inhaled GM-CSF or rituximab) is recommended1. In our case, it is worth clarifying that it was unnecessary to perform complete lung lavage after bronchoscopy, as a case with a self-limiting course.
CONCLUSION
Despite PAP being a rare condition, it should be considered in the differential diagnosis in cases of suspected persistent pneumonia despite initial management. We presented a case of PAP associated with adenovirus infection, an unusual presentation. The role of possible viral causation ‘trigger’ of PAP in cases of adenovirus infection needs to be studied.